

Two cases of 16p11.2 deletion syndrome in adolescent girls with genital malformations
Tsabai P.N., Mukosey I.S., Batyrova Z.K., Pavlova N.S., Kumykova Z.Kh., Sadelov I.O., Kirillova I.I., Voskoboinikov A.A., Shubina J., Uvarova E.V.
Background: Chromosome 16p11.2 deletion syndrome is a clinically heterogeneous condition characterized by impaired psychomotor development, obesity, and mental disorders. The disorder has an autosomal dominant inheritance pattern; however, due to incomplete penetrance and variable manifestations, microdeletion carriage often goes unnoticed. The frequency of 16p11.2 deletions among patients with developmental delays, autism spectrum disorders, schizophrenia, obesity, and genitourinary malformations is several times higher than in the general population. In Mayer–Rokitansky–Küster–Hauser syndrome, 16p11.2 microdeletions involving the TBX6 gene are a common genetic finding. Point variants in TBX6 gene have been described in Herlyn–Werner–Wunderlich syndrome.
Objective: To study the genetic causes of uterine and vaginal anomalies in two patients with the 16p11.2 microdeletion.
Materials and methods: Whole-exome sequencing with copy number variation analysis was performed in the patients. Patient G. also underwent chromosomal microarray analysis. Low-coverage genome sequencing was performed in the patients and their parents to determine the origin of the 16p11.2 deletion.
Results: A proximal 16p11.2 deletion was detected in patient E. with uterine and vaginal aplasia and in patient G. with Herlyn–Werner–Wunderlich syndrome. The patients had the following extragenital symptoms of the genetic disorder: thrombocytopenia, obesity, and skeletal anomalies were noted in patient E.; a delay in psychomotor development, renal aplasia, and skeletal anomalies were revealed in patient G.
Conclusion: These cases confirm the pleiotropic effects of the 16p11.2 deletion and the need for a multidisciplinary approach to patients with this genetic variant. Given the absence of pathognomonic features, differential diagnosis with this disease should be considered among patients with genitourinary anomalies, delayed psychomotor development, obesity, and hematological anomalies. Since these patients are reproductively capable, identifying the 16p11.2 microdeletion significantly impacts the risk assessment for congenital pathologies in their children and pregnancy planning strategies.
Authors’ contributions: Tsabai P.N., Mukosey I.S., Shubina J. – developing the concept and design of the study; Tsabai P.N., Batyrova Z.K., Kumykova Z.Kh. – consulting the patients, collecting and processing the material; Mukosey I.S., Sadelov I.O., Kirillova I.I., Voskoboinikov A.A. – bioinformatics analysis; Tsabai P.N., Mukosey I.S., Pavlova N.S., Batyrova Z.K. – writing the text; Shubina J., Uvarova E.V. – editing the article.
Conflicts of interest: Authors declare lack of the possible conflicts of interest.
Funding: This work was supported by the Russian Science Foundation, Grant No. 25-65-00040.
Ethical Approval: This study was approved by the Ethics Committee (Minutes No. 07 of the meeting of the Biomedical Research Ethics Committee at the V.I. Kulakov National Medical Research Center of the Ministry of Health of Russia dated 21.08.2025).
Patient Consent for Publication: The patients and their parents provided an informed consent to the use of all information for scientific purposes.
Authors' Data Sharing Statement: The data supporting the findings of this study are available on request from the corresponding author after approval from the principal investigator.
For citation: Tsabai P.N., Mukosey I.S., Batyrova Z.K., Pavlova N.S., Kumykova Z.Kh., Sadelov I.O.,
Kirillova I.I., Voskoboinikov A.A., Shubina J., Uvarova E.V. Two cases of 16p11.2 deletion syndrome in
adolescent girls with genital malformations.
Akusherstvo i Ginekologiya/Obstetrics and Gynecology. 2025; (12): 112-119 (in Russian)
https://dx.doi.org/10.18565/aig.2025.315
Keywords
16p11.2 microdeletion
TBX6
whole-exome sequencing
chromosomal microarray analysis
genital malformations
Mayer–Rokitansky–Küster–Hauser syndrome
Herlyn–Werner–Wunderlich syndrome
OHVIRA
scoliosis
developmental delay
Deletion of the chromosomal region 16p11.2 is one of the most common genome copy number variations in the population (carrier frequency of 1:3100 individuals) [1]. It occurs due to nonallelic homologous recombination of low-copy repeats in this region. However, due to incomplete penetrance and variability in clinical manifestations, carriage of this microdeletion often goes unnoticed. Various studies estimate the penetrance of proximal 16p11.2 deletions involving the TBX6 gene at 18–47% [2, 3].
Chromosome 16p11.2 deletion syndrome is a clinically heterogeneous condition characterized by impaired psychomotor and intellectual development, hyperphagia, obesity and mental disorders. The disorder has an autosomal dominant inheritance pattern with a 50% probability of transmission regardless of gender. The frequency of 16p11.2 deletions among patients with developmental delay, autism spectrum disorders, schizophrenia, obesity and genitourinary malformations is several times higher than in the general population [4]. It is still unknown what factors determine the penetrance and expressivity of this pleiotropic genetic variation.
Among patients with uterine and vaginal aplasia, or Mayer-Rokitansky-Küster-Hauser syndrome (MRKH), 16p11.2 microdeletions involving the candidate gene TBX6 are a common genetic finding [5]. After the identification of the TBX6 gene as a key gene for genitourinary development, single nucleotide variants in TBX6 were described in müllerian agenesis syndrome without the 16p11.2 microdeletion and in other female genital anomalies: distal vaginal atresia, obstructed hemivagina-unilateral renal agenesis (OHVIRA, also known as Herlin-Werner-Wunderlich syndrome) [6, 7].
The article presents a description of two patients with uterine and vaginal anomalies and the 16p11.2 microdeletion.
Materials and methods
This study was approved by the Ethics Committee. The study complies with the World Medical Association Declaration of Helsinki. The patients and their parents provided an informed consent to the use of all information for scientific purposes. The biological material (blood) was collected in EDTA-containing tubes, and DNA extraction was performed using PREP-MB MAX DNA Extraction Kit (DNA-Technology) or ExtractDNA Blood & Cells (Eurogen) according to the manufacturer’s protocol. Chromosomal microarray analysis was performed using CytoScan 750K kits (Thermofisher) according to the manufacturer’s protocol. The libraries for whole-exome sequencing were prepared using the Whole-Genome Library Preparation Reagent Kit ((DNA-Technology), and enrichment with target fragments was performed using the VAHTS Target Capture Core Exome Panel kit (Vazyme). Sequencing of the enriched fragments was performed on a NovaSeq 6000 sequencer. Sequencing data processing was performed using an algorithm that included read alignment to the hg38 reference genome sequence, calling and variant quality filtering. All variants that passed quality filtering were annotated using the Ensembl Variant Effect Predictor (VEP) and a number of variant significance prediction algorithms (SIFT, PolyPhen-2, SpliceAI). The Genome Aggregation Database (gnomAD) was used to estimate the population frequencies of the identified variants. The OMIM, ClinVar, LOVD, and other specialized databases (where available), as well as literature data, were used to assess the clinical relevance of the identified variants. The data were interpreted in accordance with ACMG recommendations for assessing the clinical significance of the results.
Additionally, low-coverage genome sequencing was performed. The patient’s DNA was sequenced using the NextSeq 550 System. The sequencing data were processed using an algorithm that included read alignment to the hg38 reference genome sequence and a search for copy number abnormalities in whole chromosomes and chromosome fragments.
Results
Case 1
Patient E. was born from the second pregnancy (mother’s first pregnancy was missed miscarriage) and first full-term normal delivery. At birth, her body weight was 3010 g, length was 52 cm, was Apgar score was 8/9. She grew and developed according to her age and was followed by a neurologist for muscle hypertonia and toe walking until she was one-year-old. As a child, she had recurrent respiratory infections. From the age of 9, she experienced occasional spontaneous epistaxis. A routine examination at age 10 revealed thrombocytopenia 90,000/μL. Subsequently, her platelet count remained within the normal range, with moderate thrombocytopenia of 50,000–80,000/μL. She was diagnosed with immune thrombocytopenia by a hematologist; no specific therapy was prescribed. Since the age of 11, patient E. has been overweight. The endocrinologist diagnosed her with grade I exogenous constitutional obesity that is not sensitive to lifestyle modification and increased physical activity.
At the age of 11, an ultrasound scan of the cervical lymph nodes revealed a rounded isoechoic lesion in the left lobe of the thyroid gland, measuring 2.0×1.9×2.0 cm with increased blood flow, and an enlargement of the submandibular lymph nodes to 2.5 cm. A thyroid ultrasound examination revealed a cystic lesion with a large vascularized component in the left lobe, measuring 9.6 cm3 and classified as TI-RADS III. Contrast-enhanced CT scan of the chest and neck revealed a cystic-solid lesion in the left lobe of the thyroid gland, measuring 40×22×24 mm, intensively accumulating contrast. Surgical treatment included a left hemithyroidectomy with isthmusectomy. Histological analysis showed a follicular adenoma of the thyroid gland, oncocytic variant.
In addition to the underlying disease, the patient complained of nosebleeds, poor posture, decreased visual acuity, stereotypical finger movements (clenching fists, finger fiddling), restless legs, tossing and turning during sleep. A comprehensive examination and consultations with specialists were conducted at the hospital. According to the hematologist, the examination results were consistent with a diagnosis of chronic immune thrombocytopenia; no therapy was necessary. The orthopedist made a diagnosis of connective tissue dysplasia syndrome (hypermobility of large and small joints). The specialist revealed scoliotic posture, grade II scoliosis, hallux valgus, transverse flatfoot, basal syndactyly of the second and third toes of both feet. The ophthalmologist diagnosed the patient with mild myopia and myopic astigmatism. The neurologist revealed simple motor stereotypies; no correction was required.
At the age of 14, she consulted a gynecologist regarding the absence of menstruation. The examination revealed aplasia of the uterus and vagina. She was hospitalized for a course of complex non-surgical colpopoiesis procedures. At the time of examination, she was in high school, her academic performance was good, and she had no difficulties communicating with her peers. On admission to hospital, her height was 159 cm and her weight was 80 kg (BMI was 32kg/m2). She took L-thyroxine 50 mcg daily.
The geneticist suspected a syndromic genital malformation. Phenotype of the patient: proportionate, hypersthenic, female-type body build. Shoe size was 37. The patient was overweight, had acne, small bruises on the back of her thighs, partial cutaneous syndactyly of toes 2–3, hypoplasia of the distal phalanges of feet. Family history was unremarkable.
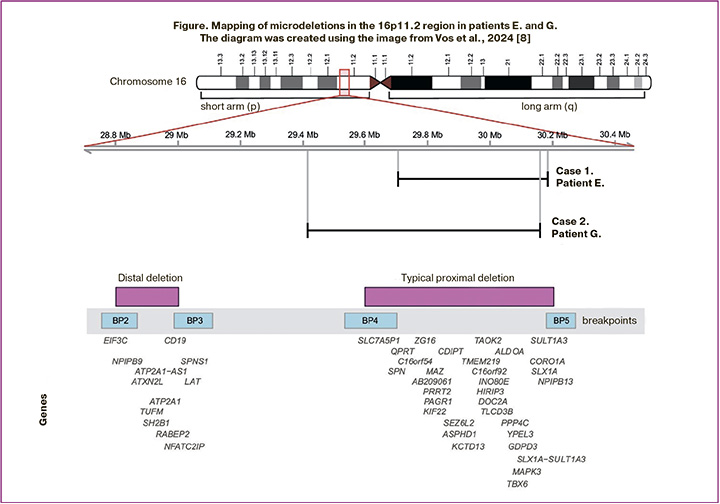
A cytogenetic study of blood lymphocytes revealed a normal female karyotype (46,XX). Whole-exome sequencing was performed to identify genetic variants that could explain patient E.’s phenotype (read depth: 113x, 10x width: 0.98, average coverage of target region 16p11.2: 84x). No single-nucleotide variants were detected. A heterozygous deletion of chromosome 16p11.2 with the approximate boundaries chr16:29679097-30188588, measuring 509.5 kb, encompassing 32 genes, including 26 protein-coding genes, was detected (Figure). The variant was registered in the gnomAD CNVs v4.1.0 control sample, comprising 114 mutant alleles on 464,284 chromosomes (homozygotes were not registered). The identified deletion was considered pathogenic.
To perform the segregation analysis, low-coverage whole-genome sequencing (read depth: 17x; average coverage of the target region 16p11.2 in the proband: 9x, in the mother: 17x) was performed on blood samples obtained from the patient and her mother (the father was not available for testing): this deletion was not inherited from the mother.
Case 2
Patient G. was born from the third pregnancy, which was a threatened miscarriage at 5 weeks’ gestation, and a second vaginal delivery at term. At birth, she weighed 3200 g, measured 50 cm in length, and had an Apgar score of 7/9. Until the age of 1 year, she developed normally, but subsequently showed delays in psychoverbal development. She was followed for the diagnosis of sequelae of organic CNS damage. At the age of 5, she was diagnosed with mixed psychoverbal development disorder. As a child, she suffered from obstructive bronchitis and varicella; she underwent cryosurgery for hemangiomas on her back, and was diagnosed with myopic astigmatism and thoracic scoliosis. Patient G. has been disabled since age 12 due to mild intellectual disability.
Menarche occurred at the age of 12, with heavy and painful periods lasting 4-15 days. At the age of 16, patient G. was admitted to local hospital due to abnormal uterine bleeding. The examination revealed a duplication of the uterus. Hemostatic therapy was administered.
She was hospitalized six months after the described episode of bleeding for examination and treatment. On admission, the patient’s physical and sexual development were appropriate for her age. Her height was 160 cm, weight was 53 kg (BMI was 21 kg/m2). A clinical and instrumental examination was performed. The ultrasound assessment of the pelvic organs (day 5 of the menstrual cycle) revealed a complete duplication of the uterus and atresia of the lower third of the vagina on the left. There was hematocolpos, hematocervix, hematometra on the left. It also showed indirect echo signs of adhesions. The renal ultrasound revealed agenesis of the left kidney for the first time and lumbar dystopia of the solitary right kidney. An MRI of the pelvic organs was performed, revealing malformations of the internal genital organs: complete duplication of the uterus with a common medial wall, duplication of the cervix and vagina with aplasia of the distal third of the vagina. Left-sided hematocervix, hematocolpos, and hematosalpinx were present. Pathological changes in the structure of the right fallopian tube should be differentiated between chronic salpingitis and reactive changes.
The combination of developmental anomalies of the reproductive and urinary systems in patient G. is consistent with Herlin-Werner-Wunderlich syndrome (OHVIRA). Surgical treatment was performed: opening and emptying the hematocolpos, creating an oval window between the closed and functioning vagina.
The geneticist suspected a syndromic form of developmental delay with anomalies of the internal genital organs. Phenotype: slanted palpebral fissures, retrognathia, slightly open mouth, malocclusion, dental caries, and thoracolumbar scoliosis. Family history: older brother and parents are healthy; mother has a bicornuate uterus.
A cytogenetic study of blood lymphocytes revealed a karyotype of 46,XX – a normal female karyotype. To search for variants explaining developmental delay and genitourinary anomaly, patient G. underwent whole-exome sequencing (read depth: 114x, 10x width: 0.98, average coverage of the target region 16p11.2: 84x). The results revealed the presence of a previously identified deletion of a region of chromosome 16 with approximate boundaries 16:29663729-30188964, 16p11.2 (hg38) of approximately 525 kbp in size. No additional variants explaining the patient’s phenotype were detected. High-resolution chromosomal microarray analysis (750K) was performed. The analysed sample with a female genotype detected a pathogenic microdeletion 16p11.2 of more than 700 kbp in size on the short arm of chromosome 16. ([GRCh38] 16:29417211-30178708) involving 31 protein-coding genes (Figure).
To conduct segregation analysis, low-coverage whole-genome sequencing (read depth: 17x; average coverage of the target region 16p11.2 in the proband: 12x, in the parents: 18-19x) was performed on blood samples obtained from patient G. and her parents. This deletion was not inherited from the parents and therefore arose de novo.
Discussion
A typical or proximal (OMIM#611913) BP4-BP5 microdeletion (29.6–30.2 Mb; ~600 kb) and a rare distal (OMIM#613444) BP2-BP3 microdeletion (28.8–29.0 Mb; ~220 kb) have been characterized in the 16p11.2 region. In both cases, the patients had a proximal 16p11.2 deletion that was not inherited from their parents. This deletion is flanked by highly homologous regions of low-copy repeats, which can lead to copy number changes through nonallelic homologous recombination [9]. Copy number variations in the chromosomal region 16p11.2 BP4-5 influence different phenotypic manifestations such as the risk of autism spectrum disorders (ASD), developmental delay and intellectual disability, schizophrenia and other mental illnesses, seizure disorders, body mass index, and head circumference. Patients with deletions and duplications in this region have opposite manifestations [4]. About 1% of patients with ASD are carriers of the 16p11.2 BP4-5 deletion, while carriers of the 16p11.2 deletion demonstrate clinical manifestations of ASD in 22% of cases, attention deficit hyperactivity disorder in 30% of cases, and 70% of deletion carriers have some kind of disorder associated with delayed speech development [9]. Candidate genes for ASD and developmental delays in this chromosomal region include MAZ, SEZ6L2, TAOK2, KCTD13, MAPK3, MVP, DOC2A, TMEM219, and CORO1A [4, 10]. Despite complete overlap in deleted clinically significant genes, the patients had significantly different phenotypes. The clinical manifestations of the patients and their representation in samples of patients with a typical 16p11.2 deletion are compared and presented in Table. These cases confirm the pleiotropic effect of the 16p11.2 BP4-5 deletion and the need for a multidisciplinary approach to patients with this genetic variant. Chung and Hererra suggested how a paediatrician or paediatric endocrinologist should manage a child diagnosed with 16p11.2 deletion, based on what is known as of 2023 [11]. Below, we will consider the molecular basis for the formation of patient phenotypes.
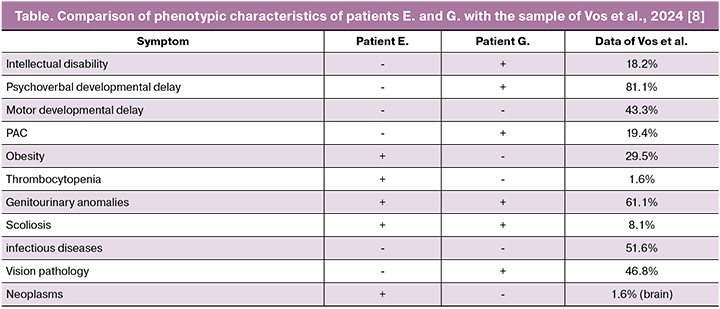
In a large study by Vos et al., including 62 Danish patients with a typical 16p11.2 microdeletion [8], genitourinary anomalies were detected in 61.1% of patients (including two patients with renal agenesis), intellectual disability was observed in 18.2%, and obesity was observed in 29.5%. Immune thrombocytopenia was also observed in one case, and nosebleeds, similar to the symptoms of patient E, were also observed in one case. Thrombocytopenia in patients with 16p11.2 microdeletions may be associated with haploinsufficiency of the CORO1A gene. The CORO1A gene encodes a cytoskeletal protein involved in the functioning of immune cells and platelets, and is believed to be associated with thrombocytopenia in patients with 16p11.2 deletions. It is worth noting that the presence of a 16p11.2 deletion may increase the risk of recessive diseases in this region in the presence of point variants in genes on the second allele. Thus, CORO1A-associated immunodeficiency has been described in patients with a 16p11.2 microdeletion, including this gene, in a compound heterozygous state with a CORO1A missense mutation [12, 13]. The patients we observed had no clinical or laboratory signs of immunodeficiency, and no variants were detected in the genes in the deletion region on the second allele.
Patient E.’s phenotype was consistent with type 2 müllerian agenesis syndrome, which, in addition to uterine and vaginal aplasia, is characterized by skeletal and renal defects, hearing impairment, and cardiac defects [14]. This phenotype is believed to be associated with haploinsufficiency of the TBX6 gene, which is involved in the development of paraxial mesoderm [15]. To date, numerous cases of müllerian agenesis types 1 and 2 have been described, both with 16p11.2 deletions and with point variants in TBX6 [5]. Interestingly, point heterozygous and compound heterozygous variants in TBX6 have also been described in patients with other genital anomalies, such as distal vaginal atresia and OHVIRA syndrome [6, 7]. In our patients, whole-exome sequencing did not detect any additional variants on the second allele. Thus, the case of patient G. confirms the association of 16p11.2 microdeletion and TBX6 haploinsufficiency with a variety of uterine anomalies, including duplication in OHVIRA syndrome.
Variants in the TBX6 gene also lead to congenital scoliosis and spondylocostal dysostosis. Congenital or idiopathic scoliosis is observed in some patients with 16p11.2 deletion [16]. Our both patients also had scoliotic spinal deformity. Some authors suggest that incomplete penetrance of disease symptoms, such as scoliosis and renal malformations, is associated with the requirement for a specific hypomorphic haplotype on the second allele without a deletion for disease manifestation [17], however, no association with this haplotype has been shown for MRKH syndrome [18]. However, this region remained uncovered by whole exome sequencing in patients E. and G. In general, 16p11.2 deletions are detected more frequently in patients with type II MRKH syndrome (4/112), but not in patients with isolated aplasia of the uterus and vagina (type I MRKH syndrome, 1/70-2/84) [19, 20]; these findings are consistent with our observations. Other candidate genes for genital anomalies in patients with typical 16p11.2 deletions include KCTD13, which encodes a protein with a tetramerization domain of potassium channels 13 [21], and MAZ, which encodes a transcription factor with a C2H2 zinc finger domain and is involved in multiple signalling pathways, including the Wnt pathway [22]. The studies of the functions of the KCTD13 and MAZ genes in mouse models indicate that haploinsufficiency of one or the other gene leads to developmental anomalies of the genitourinary system in males, including unilateral renal agenesis, but there are no reports of uterine anomalies in females. Thus, TBX6 is currently the key gene, and its deletion in 16p11.2 microdeletion syndrome causes uterine and vaginal anomalies.
In addition to the association of proximal 16p11.2 deletion with female reproductive system defects, the patients with this syndrome have been reported to have cryptorchidism [8,10], hypospadias [11], partial insensitivity to androgens, glucocorticoids, and thyroid hormones [12], precocious puberty [13], and a case of a rare tumour, pituitary germinoma, which led to hypogonadotropic hypogonadism, hypothyroidism, diabetes insipidus, and secondary adrenal insufficiency [14].
It is interesting to note that the deletions identified in these patients include the PRRT2 gene. Heterozygous variants in this gene lead to several neurological disorders: benign familial infantile seizures, paroxysmal kinesigenic dyskinesia (PKD), PKD with infantile seizures, and familial hemiplegic migraine [23]. In the case of patient E., stereotypical finger movements were observed under stress, but they did not resemble athetosis. None of the patients had a personal or family history of seizure disorders. Therefore, it cannot be reliably concluded that the patients had symptoms of PKD.
Overweight is observed in most adult patients with 16p11.2 deletions. Obesity is characteristic of both proximal and distal deletions. The major gene regulating energy balance in distal deletions is SH2B1, whereas in proximal deletions, the deletion of microRNA genes is thought to affect the development of the hypothalamus and the leptin-melanocortin pathway [24]. Despite the high penetrance of the deletion in relation to overweight, obesity was observed only in patient E., while patient G. had normal body weight.
Conclusion
Given the absence of pathognomonic features for 16p11.2 deletion syndrome, differential diagnosis with this disorder should be considered in patients with anomalies of the reproductive system and urinary tract, delayed psychomotor and intellectual development, obesity, and hematological anomalies. Since these patients typically have intact ovarian reserve and reproductive potential, identifying a 16p11.2 microdeletion significantly impacts the risk assessment for congenital pathologies in their children and pregnancy planning strategies.
References
- Smajlagić D., Lavrichenko K., Berland S., Helgeland Ø., Knudsen G.P., Vaudel M. et al. Population prevalence and inheritance pattern of recurrent CNVs associated with neurodevelopmental disorders in 12,252 newborns and their parents. Eur. J. Hum. Genet. 2021. 29(1): 205-15. https://dx.doi.org/10.1038/s41431-020-00707-7
- Goh S., Dudding-Byth T., Pinese M., Kirk E.P. Updated penetrance estimates for recurrent copy number variants – an improved definition and formula. Eur. J. Hum. Genet. 2025; 26: 34. https://dx.doi.org/10.1038/s41431-025-01948-0
- Goh S., Thiyagarajan L., Dudding-Byth T., Pinese M., Kirk E.P. et al. A systematic review and pooled analysis of penetrance estimates of copy-number variants associated with neurodevelopment. Genet. Med. 2025; 27(1): 101227. https://dx.doi.org/10.1016/j.gim.2024.101227
- Auwerx C., Kutalik Z., Reymond A. The pleiotropic spectrum of proximal 16p11.2 CNVs. Am. J. Hum. Genet. 2024; 111(11): 2309-46. https://dx.doi.org/10.1016/j.ajhg.2024.08.015
- Herlin M.K. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: advancements and implications. Front. Endocrinol. 2024; 15: 1368990. https://dx.doi.org/10.3389/fendo.2024.1368990
- Chu C., Li L., Lu D., Duan A.H., Luo L.J., Li S. et al. Whole-exome sequencing identified a TBX6 loss of function mutation in a patient with distal vaginal atresia. J. Pediatr. Adolesc. Gynecol. 2019; 32(5): 550-4. https://dx.doi.org/10.1016/j.jpag.2019.06.006
- Tewes A.C., Hucke J., Römer T., Kapczuk K., Schippert C., Hillemanns P. et al. Sequence variants in TBX6 are associated with disorders of the müllerian ducts: an update. Sex. Dev. 2019; 13(1): 35-40. https://dx.doi.org/10.1159/000496819
- Vos N., Kleinendorst L., van der Laan L., van Uhm J., Jansen P.R., van Eeghen A.M. et al. Evaluation of 100 Dutch cases with 16p11.2 deletion and duplication syndromes; from clinical manifestations towards personalized treatment options. Eur. J. Hum. Genet. 2024; 32(11): 1387-1401. https://dx.doi.org/10.1038/s41431-024-01601-2
- Chung W.K., Roberts T.P., Sherr E.H., Snyder L.G., Spiro J.E. 16p11.2 deletion syndrome. Curr. Opin. Genet. Dev. 2021; 68: 49-56. https://dx.doi.org/10.1016/j.gde.2021.01.011
- Leone R., Zuglian C., Brambilla R., Morella I. Understanding copy number variations through their genes: a molecular view on 16p11.2 deletion and duplication syndromes. Front. Pharmacol. 2024; 15: 1407865. https://dx.doi.org/10.3389/fphar.2024.1407865
- Chung W.K., Herrera F.F. Health supervision for children and adolescents with 16p11.2 deletion syndrome. Cold Spring Harb. Mol. Case Stud. 2024; 9(4): a006316. https://dx.doi.org/10.1101/mcs.a006316
- Khoreva A., Butov K.R., Nikolaeva E.I., Martyanov A., Kulakovskaya E., Pershin D. et al. Novel hemizygous CORO1A variant leads to combined immunodeficiency with defective platelet calcium signaling and cell mobility. J. Allergy Clin. Immunol. Glob. 2023; 3(1): 100172. https://dx.doi.org/10.1016/j.jacig.2023.100172
- Stocker T.J., Pircher J., Skenderi A., Ehrlich A., Eberle C., Megens R.T.A. et al. The actin regulator coronin-1A modulates platelet shape change and consolidates arterial thrombosis. Thromb. Haemost. 2018; 118(12): 2098-111. https://dx.doi.org/10.1055/s-0038-1675604
- Кругляк Д.А., Буралкина Н.А., Ипатова М.В., Батырова З.К., Уварова Е.В. Аплазия влагалища и матки (синдром Майера-Рокитанского-Кюстнера-Хаузера): этиология, патогенетические аспекты и теории формирования порока (обзорлитературы). Гинекология. 2018; 20(2): 64-6. [Kruglyak D.A., Buralkina N.A., Ipatova M.V., Batyrova Z.K., Uvarova E.V. Aplasia of the vagina and uterus (Mayer-Rokitansky-Kustner-Hauser syndrome): etiology, pathogenetic aspects and theory of the formation of defect (literature review). Gynecology. 2018; 20(2): 64-6 (in Russian)]. https://dx.doi.org/10.26442/2079-5696_2018.2.64-66
- Nik-Zainal S., Strick R., Storer M., Huang N., Rad R., Willatt L. et al. High incidence of recurrent copy number variants in patients with isolated and syndromic Müllerian aplasia. J. Med. Genet. 2011; 48(3): 197-204. https://dx.doi.org/10.1136/jmg.2010.082412
- Chen W., Liu J., Yuan D., Zuo Y., Liu Z., Liu S. et al. Progress and perspective of TBX6 gene in congenital vertebral malformations. Oncotarget. 2016; 7(35): 57430-41. https://dx.doi.org/10.18632/oncotarget.10619
- Liu J., Wu N., Yang N., Takeda K., Chen W., Li W., Du R. et al. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet. Med. 2019; 21(7): 1548-58. https://dx.doi.org/10.1038/s41436-018-0377-x
- Ma C., Chen N., Jolly A., Zhao S., Coban-Akdemir Z., Tian W. et al. Functional characteristics of a broad spectrum of TBX6 variants in Mayer-Rokitansky- Küster-Hauser syndrome. Genet. Med. 2022; 24(11): 2262-73. https://dx.doi.org/10.1016/j.gim.2022.08.012
- Su K., Liu H, Ye X, Jin H, Xie Z, Yang C. et al. Recurrent human 16p11.2 microdeletions in type I Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome patients in Chinese Han population. Mol. Genet. Genomic Med. 2024; 12(1): e2280. https://dx.doi.org/10.1002/mgg3.2280
- Chen N., Zhao S., Jolly A., Wang L., Pan H., Yuan J. et al. Perturbations of genes essential for Müllerian duct and Wölffian duct development in Mayer-Rokitansky-Küster-Hauser syndrome. Am. J. Hum. Genet. 2021; 108(2): 337-45. https://dx.doi.org/10.1016/j.ajhg.2020.12.014
- Seth A., Rivera A., Chahdi A., Choi I.S., Medina-Martinez O., Lewis S. et al. Gene dosage changes in KCTD13 result in penile and testicular anomalies via diminished androgen receptor function. FASEB J. 2022; 36(11): e22567. https://dx.doi.org/10.1096/fj.202200558R
- Haller M., Au J., O'Neill M., Lamb D.J. 16p11.2 transcription factor MAZ is a dosage-sensitive regulator of genitourinary development. Proc. Natl. Acad. Sci. USA. 2018; 115(8): E1849-58. https://dx.doi.org/10.1073/pnas.1716092115
- Sen K., Genser I., DiFazio M., DiSabella M. Haploinsufficiency of PRRT2 leading to familial hemiplegic migraine in chromosome 16p11.2 deletion syndrome. Neuropediatrics. 2022; 53(4): 279-82. https://dx.doi.org/10.1055/a-1863-1798
- da Silva Assis I.S., Salum K.C.R., Felício R.F.M., Palhinha L., de Medeiros Abreu G., Silva T. et al. Genomic deletions on 16p11.2 associated with severe obesity in Brazil. Front. Endocrinol. (Lausanne). 2025; 15: 1495534. https://dx.doi.org/10.3389/fendo.2024.1495534
Received 30.10.2025
Accepted 09.12.2025
About the Authors
Polina N. Tsabai, geneticist, MD, Department of Clinical Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, +7(916)940-22-06, polinatsabai@gmail.com,https://orcid.org/0000-0001-5110-0827
Irina S. Mukosey, Researcher, Laboratory of Genomic Data Analysis, Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997,
https://orcid.org/0000-0002-2225-8366
Zalina K. Batyrova, gynecologist, MD, PhD, Department of Children and Adolescent Gynecology, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, linadoctor@mail.ru,
https://orcid.org/0000-0003-4997-6090
Nadezda S. Pavlova, Junior Researcher, Department of Clinical Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, pavnadser@gmail.com,
https://orcid.org/0000-0001-5619-2695
Zaira Kh. Kumykova, gynecologist, MD, PhD, Department of Children and Adolescent Gynecology, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, zai-kumykova@yandex.ru, https://orcid.org/0000-0001-7511-1432
Igor O. Sadelov, geneticist, Laboratory of Genomic Data Analysis, Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, sadelovigor@gmail.com, https://orcid.org/0000-0002-5144-6307
Irina I. Kirillova, specialist, Laboratory of Genomic Data Analysis, Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997,
https://orcid.org/0009-0008-2182-9631
Alexander A. Voskoboinikov, Junior Researcher, Laboratory of Genomic Data Analysis, Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997,
https://orcid.org/0000-0002-4543-7156
Jekaterina Shubina, PhD, Head of Laboratory of Genomic Data Analysis, Institute of Reproductive Genetics, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, e_shubina@oparina4.ru, https://orcid.org/0000-0003-4383-7428
Elena V. Uvarova, Dr. Med. Sci., Professor, Corresponding Member of RAS, Head of Department of Pediatric and Adolescent Gynecology, Academician V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology, and Perinatology, Ministry of Health of the Russian Federation, 4, Ac. Oparina str., Moscow, Russian Federation, 117997, elena-uvarova@yandex.ru, https://orcid.org/0000-0002-3105-5640
Corresponding author: Polina N. Tsabai, polinatsabai@gmail.com
Similar Articles

